A clonogenic assay, also known as a colony formation assay
is an in vitro cell survival assay. It assesses the ability of single cells to survive and reproduce to form colonies1. This assay was first described in the 1950s, where it was used to study the effects of radiation on cancer cell survival and growth and has subsequently played an essential role in radiobiology2.
In order to measure clonogenicity, cells need to be seeded at very low densities and left for a period of 1-3 weeks for colonies to form. Colonies are then fixed, stained with crystal violet to make them visible, and counted. Cell survival curves are plotted to analyze the data. Today, clonogenic assays are used to answer a variety of experimental questions, especially in cancer biology.
This blog highlights:
- The clonogenic survival assay
- How to perform a clonogenic assay
- Traditional protocol for clonogenic assays
- How to analyze your clonogenic assay
- Label-free, non-endpoint, semi-automated clonogenic assay protocol
The clonogenic survival assay
Clonogenic assays are widely used in the field of cancer research as the formation of clones is interpreted as a trait of cancer cells with tumor-initiating capabilities. While this assay was initially used in the field of radiobiology, it has become a standard tool in cancer research to evaluate cellular growth and the cytotoxic or genotoxic effects of various agents with potential clinical application. This includes chemotherapeutic agents and targeted therapies on their own or in combination3.
------------------------------------------------------
With CytoSMART Omni live-cell imager, you can analyze and quantify colony formation. Learn more about the Omni here.
------------------------------------------------------
Clonogenic growth is also used for evaluating the stemness of particular cell populations as stem cells are long-living cells with the potential for ongoing proliferation. This is particularly relevant to cancer research as cancer stem cells are often associated with chemoresistance, the formation of secondary tumors, and cancer recurrence. Therefore, investigating cancer-stemness using a clonogenic assay is a valuable and widely used tool to predict the efficacy of a particular therapy4.
Clonogenic assays measure the ability of cells to retain their reproductive integrity over a prolonged period of time. This is an important feature as it reveals phenotypic effects that require time and possibly several cell divisions to develop. When analyzing therapeutic resistance this is particularly important. Drug resistance cannot be identified using short-term cytotoxicity assays5.
How to perform a clonogenic assay
The information in this section is adapted from Franken et al. (2006) who published a clonogenic assay protocol in Nature Protocols1, combined with some insights from my own experience gained from performing over 60 clonogenic assays during my Ph.D.
Essentially, there are two different ways to perform a clonogenic assay:
(1) Cells can be seeded at low densities and then treated to examine the effect of the treatment on the clonogenicity of cells.
(2) Cells can be treated for a specified period and then re-plated at low densities in treatment-free media to test clonogenic ability.
The latter is often used to assess therapeutic resistance after treatment. Figure 1 summarizes the first approach which is the traditional method to perform a clonogenic assay. As it will become clear, this is a time-consuming method that provides no information on the progression of the experiment (endpoint only). We will discuss automated and non-endpoint alternative methods later.
Traditional protocol for clonogenic assays
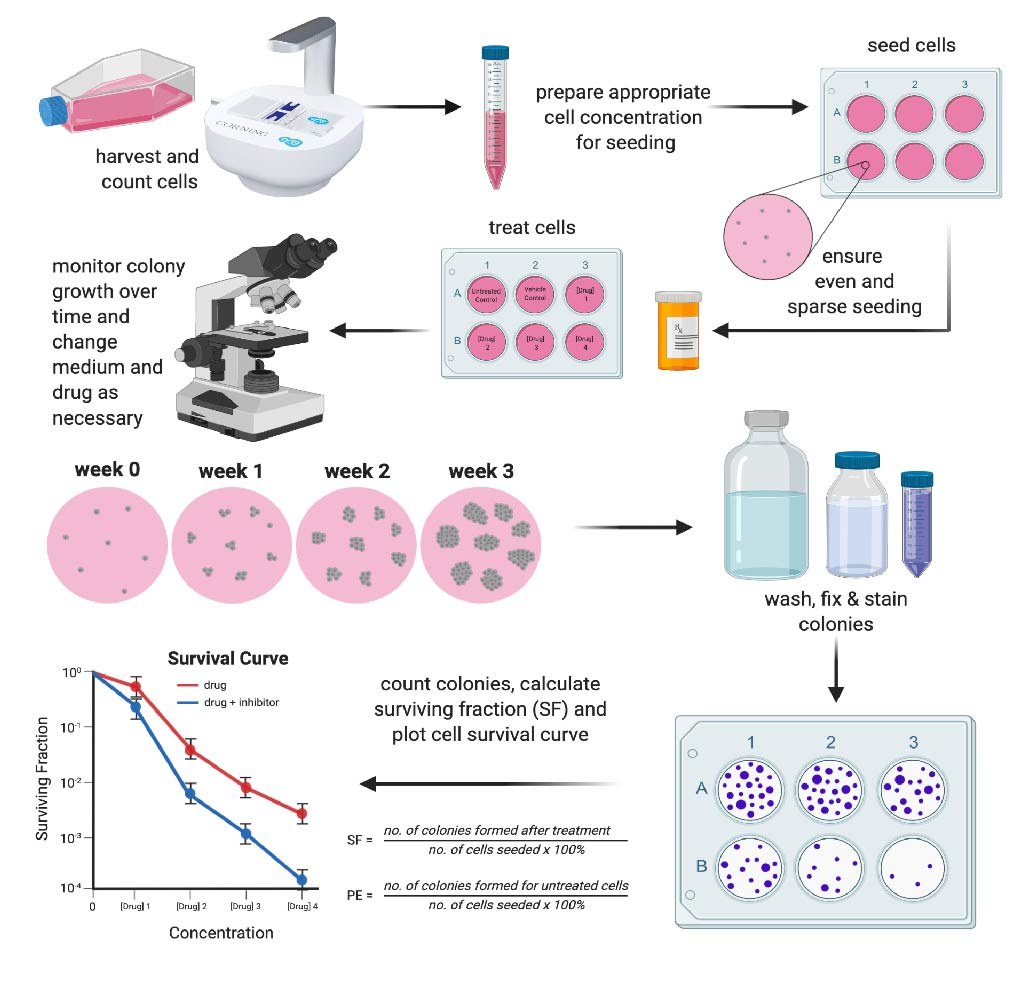
Clonogenic assays are typically performed in 6-well or 24-well plates. Cells need to be sparsely and evenly plated so isolated colonies can form. Therefore, it is important to optimize your seeding density for your chosen well size before performing your clonogenic assay.
A good starting point is to look in the literature to see if any studies have performed clonogenic assays with your cell line and then test this seeding density and two or three others. During this optimization process, it is also important to consider your experimental endpoint (e.g. 7 days, 14 days, or 21 days) and cell growth rate as you don’t want colonies to merge, which will interfere with colony quantification and analysis. Seeding densities should be the same for all treatment conditions within your experiment.
Using the right controls is critical for the analysis stage. One should always use an untreated control as well as a vehicle-only control (this could be DMSO, PBS, or whatever solvent your treatment is dissolved in). For treatment conditions, a dilution series is often used. The treatment period and harshness of your treatment will help determine your concentration range - this is likely to require some optimization. Each control and experimental condition should be performed in triplicate.
During the colony-formation phase, you will need to monitor colony growth under all treatment conditions. A colony is considered to be 50 cells or more and they are only visible under a microscope. As this assay is typically carried out over a long period of time, you will need to change your cell media. Cells will be at a very low confluence to start with, so you will not need to change the media as regularly as normal. However, if you are seeding and then treating cells with a drug or compound, it is important to consider its half-life and add fresh treatment as needed.
----------------
For researchers that are interested in imaging the colony growth over time, we recommend looking at the CytoSMART Omni.
-----------------
It is often the colonies under the control conditions that grow the fastest and therefore you can use these cells as an indication of your experimental endpoint, i.e. end the experiment before your colonies start merging and if this happens too quickly, rather use a lower seeding density for your experiment. It is important to end the experiment for all treatment conditions at the same time.
Once you have reached your experimental endpoint, cells need to be washed gently with PBS (add the PBS to the side of the well to not disrupt the colonies), fixed, and stained with the DNA intercalating dye, crystal violet (0.5% w/v) for at least 30 minutes. Removing the excess stain can be messy. The best technique is to gently dunk the plates in beakers immersed with water until all excess stain has been removed and you are only left with bright-purple colonies. Stained colonies can be counted up to 50 weeks after staining. Take high-resolution pictures of your wells to use for analysis, presentations, and publication figures.
To support researchers in the analysis and optimization of their colony formation assays, CytoSMART has introduced an application for the CytoSMART Omni to detect colonies in (time-lapse) images of entire well-plates at 10X magnification. In the incubator, time-lapse imaging can provide kinetic information rather than an endpoint, which can provide more detailed information about when the treatment starts to affect the cells. Label-free image analysis can be used to detect colonies, evaluate their size and circularity, as well as pinpoint when colonies are starting to merge and optimize the assay accordingly.
How to analyze your clonogenic assay
It really is as simple as manually counting your colonies for each treatment condition and representing the data as a survival curve (you should use at least three biological repeats for your curve).
A survival curve requires the surviving fraction (SF) of treated cells (this includes a ratio of colonies formed to cells seeded) to be calculated and plotted against the treatment dose. Before you can calculate the SF, the plating efficiency (PE) of your cells needs to be determined as different cell lines have different plating efficiencies and this affects the survival fraction calculation.
PE is the ratio of the number of colonies to the number of cells seeded in your untreated cells1. Figure 1 shows the PE and SF formulae and an example of a survival curve.
The manual counting of colonies is tiresome and can be prone to bias, so a number of freely available computerized image analysis tools have been developed for analyzing clonogenic assay images quickly and objectively. A worthy mention is the freely available software package developed by Brzozowska et al. (2019) that not only counts colonies but also plots their size distributions, which is another layer of useful cellular behavior information (Figure 2a)6.
An alternative to counting and quantifying individual colonies is determining the percentage of the well area that is covered by colonies (colony area percentage) to quantify clonogenic cell growth. Guzmán et al. (2014) developed a freely available ImageJ plugin called ColonyArea that does just this (Figure 2b)7. This is a useful tool to use if you have merged colonies that are difficult to count by eye or by colony counting software or if you want a quick and high-throughput analysis method.
Label-free, non-endpoint, semi-automated clonogenic assay protocol
A recent paper by Mayr et al. (2018) describes a new and modified clonogenic assay protocol that uses a 96-well microplate format and confluence detection to measure colonies. This method enables comprehensive experimental setups using a 96-well plate, it is label-free, has no staining endpoint, and the analysis is semi-automatic (Figure 3)8. Furthermore, time-resolved, non-endpoint confluence measurement of the same well showed that semi-automatic analysis was suitable for determining colony number and mean size.
This clonogenic assay method provides a time- and cost-effective alternative to the standard clonogenic assay protocol. With no endpoint fixation and staining required, this protocol enables continuous monitoring and analysis of clonogenic growth. Furthermore, additional metrics about the kinetics of colony growth can also be extracted during the experiment. The miniaturized format also opens up the opportunity to test expensive treatments, such as siRNA-based or CRISPR/Cas9-based therapies that would not be feasible with the volume of treatment required for a 6-well or 24-well plate. Ultimately, Mayr et al. demonstrated that label-free microscopy with confluence detection is a robust and viable option for measuring clonogenicity.
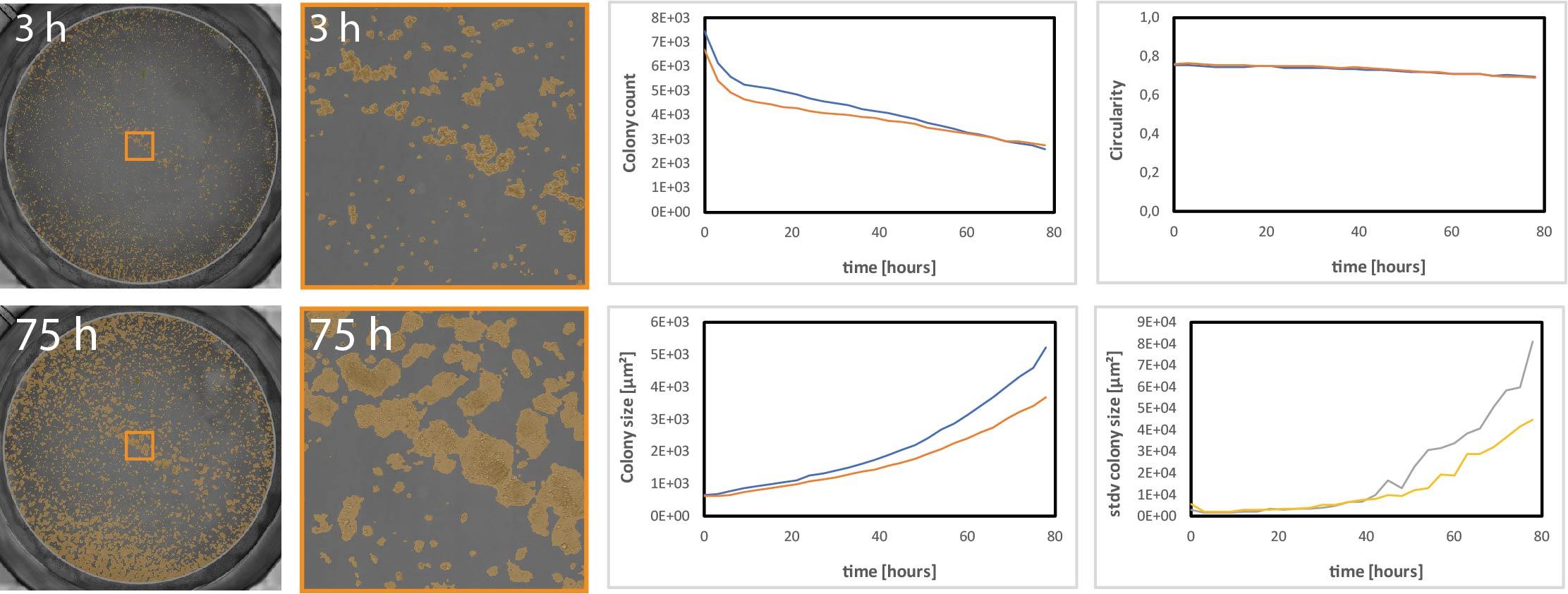
A label-free, non-endpoint, and automated solution for your clonogenic assays is the CytoSMART Omni with its colony detection algorithm. Colony formation is measured over time across an entire well with colony count, size, and circularity readouts. Your cells do not have to leave the incubator and cellular kinetic information during the process of colony formation can be obtained (Figure 4).
------------------------------------------------------
CytoSMART live-cell analysis platform enables scientists to continuously monitor colony growth over long periods of time instead of limiting data acquisition to a single time point.
Learn more about the clonogenic assay on Cytosmart imaging systems

