Compound efficacy assays that study cell death through the analysis of growth curves
Historically, most drugs that found their way to the market came from natural sources, from which the active components were extracted and identified. Many of these components were discovered serendipitously. This changed when chemistry knowledge evolved and pharmacology had become a well-defined scientific discipline.
Nowadays, drug discovery involves a more rational process that starts with the identification of candidates we want to target, followed by the synthesis, characterization, and screening in the search of active compounds for therapeutic efficacy. Compounds are screened using many different assays that elucidate affinity to the drug target and the downstream effect this has on cells. Once a compound seems like a potential drug candidate, it is tested on cell and animal models and it will begin the process of drug development in clinical trials1.
In this article, we will start by discussing different cell death assays to study cell cytotoxicity in drug discovery and we will specifically focus on a label-free method to set up kinetic assays, rather than endpoint assays.
Cell death can be measured through indirect surrogate markers using vital dyes, measuring intracellular proteins, or by determining the activity of the cellular metabolism. However, these techniques require a ‘label’ that you measure, using these introduces some inherent complications to the measurements.
A label-free method that delivers kinetic information, is to determine cell growth curves through the measurement of cell confluency, using a microscope. Generating growth curves can provide insight into cellular proliferation and apoptosis, thus different drug treatments can be easily compared in a time-dependent manner. We will explain how live-cell imaging techniques can be used to achieve this goal.
Finally, a case study of the use of growth curves in the analysis of drug effects will demonstrate the importance of this technique in drug discovery.
Cell death assays in drug development
One common feature in many human diseases is dysregulated cell death. For example, cancer is the uncontrolled growth of abnormal cells anywhere in a body. In healthy cells, programmed cell death (PCD) pathways are used to initiate apoptosis if cell damage occurs. PCD pathways are complex, cell death blockers and inducers are working in a delicate balance to achieve proper tissue homeostasis2. The modulation of this cellular mechanism has been demonstrated to be an effective therapeutic strategy for many human diseases. To understand the modulatory effect of drug candidates on PCD mechanisms, cell-based tests that quantify cell death are widely used in drug development. There are many techniques developed to assess this process, these techniques either measure cell death directly or indirectly through surrogate markers3.
Measuring cell death indirectly through surrogate markers
The most common way to quantify cell death is through the use of vital dyes. These dyes are fluorescent or colored molecules, which can be used to discriminate between living and dead cells. Exclusion dyes are compounds that are fluorescent but cannot penetrate intact membranes, hence they will only label dead cells. Alternatively, compounds that can penetrate intact membranes are used. These will only become fluorescent when they are hydrolyzed, thereby only labeling intact, living cells. However, this intracellular hydrolyzation may be affected by other unrelated phenomena besides cell death, which reduces the accuracy of the experiment and has to be quantified to determine the significance of the outcome3. Another method to quantify cell death is to look at plasma membrane rupture, by measuring the leaking of intracellular proteins into the cell culture supernatant. Although a major drawback of this technique is that the activity of these proteins (which are often enzymes) may decay over time.
Instead of quantifying cell death, compound cytotoxicity can also be analyzed by looking at cell viability. A widely used surrogate biochemical marker for cell viability is adenosine triphosphate (ATP), which is the most common energy-carrying molecule found in viable cells, thus important for cellular metabolism. Luciferase-based assays can sensitively measure this molecule and can therefore be used to quantitatively derive cell viability. However, decreased ATP concentrations may result from various cellular disruptions, which do not always directly correlate with cell viability. Another biochemical marker that is used to measure cellular viability is the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test. MTT is a colorless salt, which is readily taken up by living cells, where it is converted into a colored compound. Unfortunately, similarly to the ATP-based assay, the lack of conversion can also be due to a number of factors and thus it does not always provide direct insight into cell viability3.
While these assays that use surrogate markers are commonly used, they have limitations. Marker depletion, using too much or too little or unrelated cellular processes that involve the markers can lead to inaccurate readings. As such, these assays require multiple control experiments to properly define the deviation that is inherent to the experiment. Another major drawback of using these markers is that they can only be used to obtain endpoint measurements. Cell cultures have to be sacrificed for every analysis. This means that from the start of the culture to the point of the final measurement no information on the health of the cell culture is obtained. Thus, researchers are required to make assumptions or set up many different cultures, which are sacrificed at different time points.
Label-free cell death analysis using growth curves
A label-free method determining cell death/viability, that does not require surrogate markers, is measuring the degree of cell attachment (confluency) in the culture vessel. Adherent cell lines that contain healthy cells will form a 2D monolayer on the culture substrate, while injured cells round up and detach.
Using a microscope that can measure the area of the surface occupied by cells in a culture plate can be an indirect indicator of cell viability. Using cell attachment as a marker for cell viability offers two main advantages. Firstly, cell viability is measured non-invasively because it is label-free, thereby avoiding probe-related interference. Secondly, it allows real-time monitoring of your cell cultures and, therefore, it permits kinetic measurements of the effect of your drug on the cells3,4. Table 1 summarizes the comparison between the discussed methods of quantifying cell viability.
Table 1| Comparison of the benefits and disadvantages of surrogate markers and label-free methods to measure cellular viability3.
Vital dyesCellular metabolismExtracellular release of proteinsCell attachment
+ facilitates the identification of dead cells by visual inspection+ rapid and inexpensive+ inexpensive+ avoiding probe-relating interferences
--+ allows the study of late events and is applicable to real-time monitoring+ allows real-time monitoring
---+ label-free
- unable to discriminate between different cell death modes- cannot discriminate between cytotoxic and anti-proliferative effects- unable to discriminate between different cell death modes- unable to discriminate between different cell death modes
--- detection might be aggravated by morphological changes of dying cells-
There are numerous methods available for the quantification of cell viability. It is always important to use more than one method to exclude non-specific effects and false positives. We will now focus on the quantification of cell death using cell attachment studies. With this method, growth curves can be generated, which are of great value in the drug discovery cascade. Kinetic experiments can be set up to assess the efficacy of the drug candidate. By using kinetic measurements to obtain cellular growth curves, the effect of a treatment can be measured. Alterations in the growth rates of cell cultures are used to demonstrate whether the treatment e.g. inhibits cancer growth.
Before growth rates can be derived, we can look at the growth phases cells go through in standard culturing conditions.
The logarithmic phase in a growth curve is important for drug discovery
The growth phases of cells typically display a sigmoidal pattern of proliferation in which different phases can be distinguished (Figure 1). To start with, cell culture is initiated by seeding cells. Immediately after seeding the cells, they enter the first stage which is called the lag phase. In this stage, the cells do not divide. This process can take several hours or even days, dependent on the seeding density and cell type. At this point in time, the cells need to recover from the trypsinization, rebuild their cytoskeleton, and excrete extracellular matrix to facilitate linkage between the cells and proliferation.
After these events, the cells enter a new cell cycle, the logarithmic (log) growth phase. Here the population of cells doubles at a rate we define as the doubling time (DT), which is characteristic for each cell line. This is the phase in which the effect of drugs and chemical agents that stimulate or inhibit cell growth can be studied.
Finally, when the cell population becomes confluent, the cellular proliferation slows down and the growth rate drops close to zero, we call this the plateau phase. Hereafter, the cells don’t have enough space and nutrients anymore to grow and the cells die. This is the decline phase, where cell death is predominant and there is a reduction in viable cells5.
Thus, the logarithmic phase is important for assessing the effects of drugs. Here we can derive the doubling time, which gives a clear indication on the status of the cells and should reflect any inhibitory effect. To obtain growth curves of cell cultures, cells have to be imaged sequentially to analyze the fraction of the surface area that is covered by the cells, the confluency of the culture. These confluency measurements need to be obtained sequentially at regular time intervals to obtain an accurate view of cell growth. For this purpose, a live-cell imaging system provides a straightforward solution.
Live-cell imaging in vitro
In the early 17th century, the first optical microscopes were developed and this transformed the scientific technology for detailed physiological analysis of tissue and cell biology. Traditional microscopes are controlled manually, thus only a small number of experimental or clinical specimens can be analyzed. This has changed due to the development of fully automated microscopic acquisition platforms and the advancement of image analysis algorithms. These developments have enhanced the throughput and robustness of quantitative imaging, enabling the integration of quantitative microscopic imaging into the drug development process6.
Automation has spawned a new discipline in microscopy: high-content imaging. Over the last 10 years, high-content microscopic platforms and image analysis tools have evolved rapidly and have become integrated within the pharmaceutical and biotechnology industry.
There are several advantages of the image-based analysis of live cells in a time-resolved manner, in comparison to the old-fashioned fixed endpoint assays6:
- Using high-content imaging, a greater spatial and temporal understanding of your cell culture is obtained, providing a more complete picture of dynamic biological processes.
- Temporal analysis to study the effect of a drug allows you to quantify transient phenotypic responses.
- The appropriate time point for endpoint studies can be optimized by looking at the complete time-response of the cells towards the drug.
- Endpoint studies often result in conflicting findings due to dynamic reversible responses of the cells towards a drug. By looking at the whole time window of how cells react, these conflicting findings can be explained.
- The adaptive response of the cells towards a drug can be characterized, which allows you to determine accurate scheduling and dosing regimens of your drug.
----------
To achieve temporal high-content imaging for whole well confluency measurements: CytoSMART Omni.
----------
In the next paragraph, we will look at a case study where Gautam et al. used growth curves to analyze cytotoxic drug responses in breast cancer cells.
Case study: the analysis of cytotoxic and synthetic drug responses in triple-negative breast cancer cells
Breast cancer is cancer that develops from breast tissue. There are different types of breast cancer, which are characterized by the presence of three important receptors: estrogen receptor (ER), progesterone receptor (PR), and the human epidermal growth factor receptor 2 (HER2)7. Cancer cells that have the ER (ER+) are dependent on estrogen for their growth, so they can be treated with drugs to block the effects of estrogen8. Patients with ER+ breast cancer generally have a better prognosis.
A breast cancer type that does not display any of these receptors, triple-negative breast cancer (TNBC), is an aggressive disease and accounts for 15-20% of all breast cancer cases. While targeted treatment exists for the receptor-positive subtypes, there is no specific treatment for TNBC, resulting in a bad prognosis. Thus, there is an urgent need for better treatment options for TNBC9.
Gautam et al.9 studied the response of 16 different TNBC cell lines to 301 compounds to investigate if these compounds selectively inhibited cell growth. They used different techniques to analyze cell cytotoxicity: a resazurin cytosolic reduction-based viability assay, a cellular ATP-based viability assay, a cell membrane impermeable DNA-binding dye-based cytotoxicity assay, and cellular growth curves.
They observed that the compounds affected the cellular ATP levels considerably, but this trend was not as obvious when they investigated occurrences of cell death. Meaning that a large group of compounds inhibited cell viability, but did not induce cell death to a similar extend. This suggests that inhibition of the signal in the cell viability assays is not directly indicative of cell death.
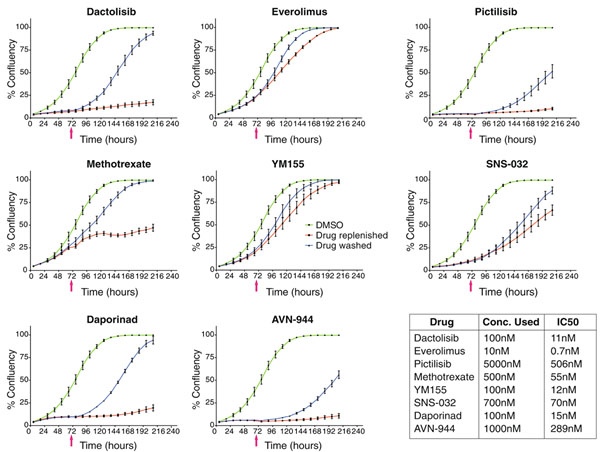
Next, they investigated whether this effect was caused by a reversible or non-reversible response to the drugs they administered. Eight different compounds that showed strong inhibition of cell viability, but that were non-toxic to the tested cell lines were selected: dactolisib, everolimus, pictilisib, methotrexate, YM155, SNS-032, daporinad, and AVN-944.
To investigate the drug reversibility, the cells were grown in the presence of the compound, and after 72 hours the compound was removed. After removal of the drug, the cells were cultured for several more days (Figure 2). They created growth curves by analyzing the confluency of microscopic images to explore the effect of the compounds on cell cytotoxicity.
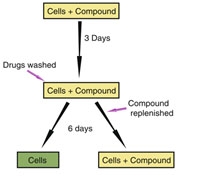
Figure 2| Schematic illustration of the experimental set-up9.
They observed that the effect of all eight compounds was reversible (Figure 3). Interestingly, in some cases, the inhibition of the drug was overcome even in the presence of the drug. In the presence of dactolisib, pictilisib, daporinad, and AVN-944, the cell growth was strongly inhibited or arrested, although the cells began dividing again after removing the compound. On the other hand, methotrexate, everolimus, YM155, and SNS-032 only caused a transient inhibitory effect. After two to five days the cells began to grow again even in the presence of these compounds. This shows that non-toxic cell viability responses are cytostatic and reversible, sometimes even in the presence of the inhibitor9.
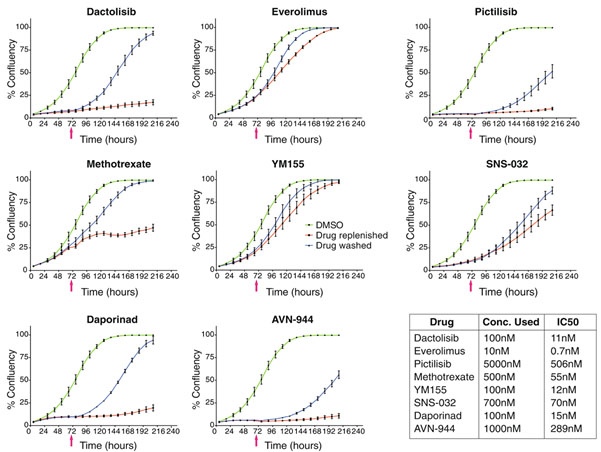
This case study shows that by studying cell death by image-based analysis of live cells in a time-resolved manner, the dynamic cellular response to certain drugs can be analyzed. By studying cell death rather than cell viability, the response observed in one time-point assay could be separated from the truly cytotoxic ones. Several drug classes could be identified that show broad cell viability readouts but didn’t show a cell-killing response. Furthermore, by studying cells in real-time using cell growth curves analysis, they were able to detect that some static responses were overcome even in the presence of the drugs. These effects couldn’t be observed with a static cell viability readout, emphasizing the importance of real-time cell analysis in drug discovery assays.
Conclusion
In this article we focused on different ways to analyze cell death, and how you can use these assays to assess compound efficacy in drug discovery. Cell attachment analysis to study cell viability is a powerful method to study the effects of drugs on a cell in a temporal manner without the use of probes which can interfere with the effects. Growth curves can be generated, where the log-phase is important for the analysis of drug-related effects.
The case study showed that by focusing on the growth of live cells in a time-resolved manner, the dynamic cellular response of the drugs could be analyzed. Effects were discovered, which would have been very difficult to explore by only using fixed time points cell death measurements. Nonetheless, it is always essential to use more than one method to assess cell viability to exclude non-specific effects and false positives. It is important to realize that by only looking at cell attachment, you cannot discriminate different cell death modes. But the technique avoids probe-related interference and it allows for real-time cell monitoring.
Related Products
There are currently no products tagged to this resource.