Gene expression profiling and gene reprogramming are two powerful tools in biomedical research. Gene expression profiling helps us identify the molecular pathways involved in physiological and pathological events. Once these pathways are identified, gene reprogramming can be utilized to alter specific genes and study their function in greater detail.
Additionally, gene reprogramming allows us to produce viral vectors, recombinant proteins, and vaccines. All of which can be used to improve our understanding and treatment of human diseases. Since both tools utilize mammalian cells, the usefulness of these technologies is entirely dependent on the quality of cell culture.
In this article, we will discuss how the proliferative rate and confluency of cells (1) affect baseline gene expression levels and, consequently, the biological interpretation of the data, and (2) influence the efficiency of gene reprogramming and the quality of the resultant products.
Cell confluency determines gene expression levels
In the fields of tissue engineering, immunology, oncology, cell metabolism, and regenerative medicine, gene expression profiling is used to delineate the molecular pathways involved in various physiological processes (Georgantas et al., 2004; Mansergh et al., 2009; Maslove and Wong, 2014; Calon et al., 2015).
In immunology, subtypes of T cells have been characterized by differences in gene expression upon infection (Wolski et al., 2017), while in regenerative medicine gene expression profiles have been used to distinguish stem cells from differentiated cells (Georgantas et al., 2004).
Gene expression profiles are generated with data collected by microarrays. A DNA microarray is a microscope slide containing thousands of tiny spots in defined positions, with each spot containing a known DNA sequence or gene (Pollack, 2009). The DNA molecules attached to each array function as probes to detect the transcriptome or the set of mRNA transcripts expressed by a group of genes.
To perform a microarray analysis, mRNA is isolated from an experimental sample and a reference sample. For example, the reference sample could be untreated cells, while an experimental sample could be cells treated with a drug. The mRNA is then reverse transcribed into complementary DNA (cDNA) and each sample is labeled with a fluorescent probe of a different color (e.g. green for reference sample, red for experimental sample). Once labeled, the samples are mixed together and hybridized to the microarray. After hybridization, the microarray is scanned to measure the expression of each gene printed on the slide. If the expression of a gene is higher in the experimental sample than in the reference sample, then the corresponding spot on the microarray fluoresces red. Conversely, if the expression in the experimental sample is lower than in the reference sample, the spot fluoresces green. However, if the expression of a gene is equal in the two samples, the spot appears yellow (Figure 1).
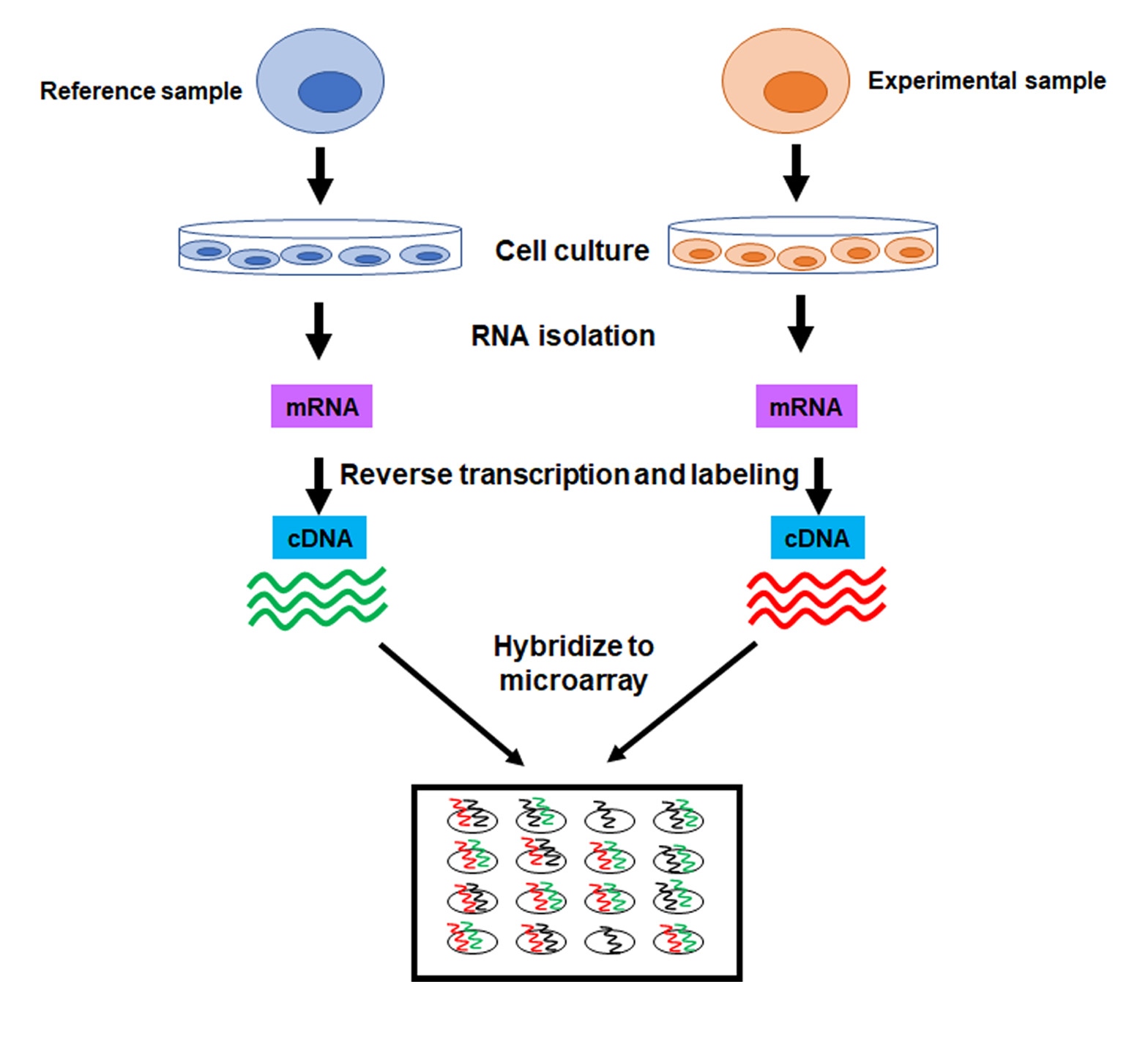
The final product of microarray analysis is a gene expression profile, which shows the expression of many genes in the experimental sample as “upregulated” or “downregulated” from the reference sample (Figure 2). Thus, it is important that the source of the DNA, the cells, of the reference sample is maintained under well-controlled conditions.
As confluency influences cell metabolism (Chacko and Eliceiri, 2019), the transcriptome of the reference sample is likely to be affected by the degree of confluency as well. Therefore, if confluency is not well controlled, the data obtained from a microarray analysis may not be interpretable or lead to incorrect conclusions.
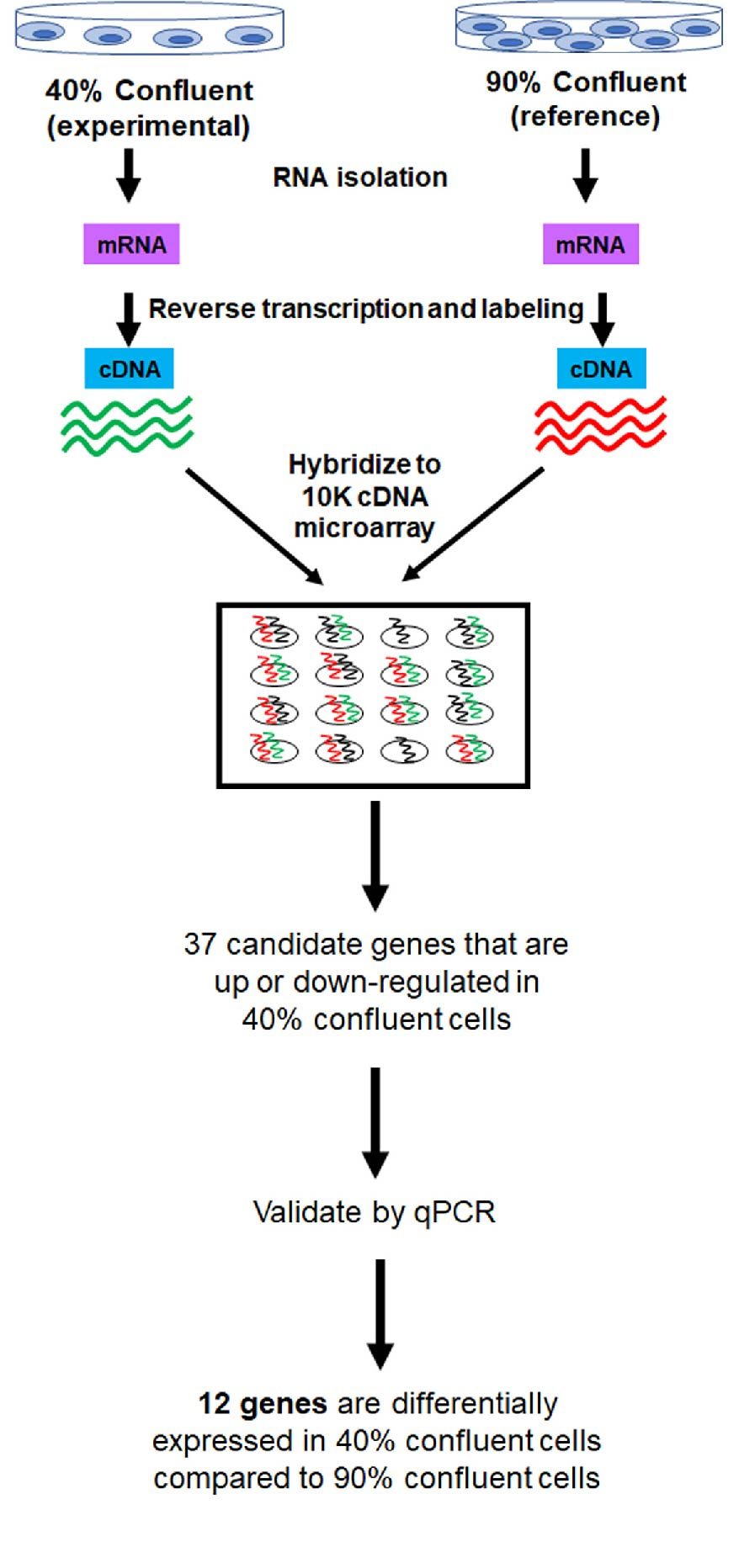
A 2006 study published by Han et al., demonstrates that confluency alters the gene expression profile of cells. Total RNA was isolated from HEK293T cells that were 40% or 90% confluent, with 90% confluent cells used as the reference sample. The isolated RNA was reverse transcribed, labeled, and hybridized to 10K cDNA microarrays. Using multivariate predictive models, the researchers identified 37 genes that were either up- or downregulated in sub-confluent (40%) cells compared to 90% confluent cells. Out of the 37 predicted genes, 12 genes were validated to be differentially expressed between the two groups by qPCR (Han et al., 2006) (Figure 3).
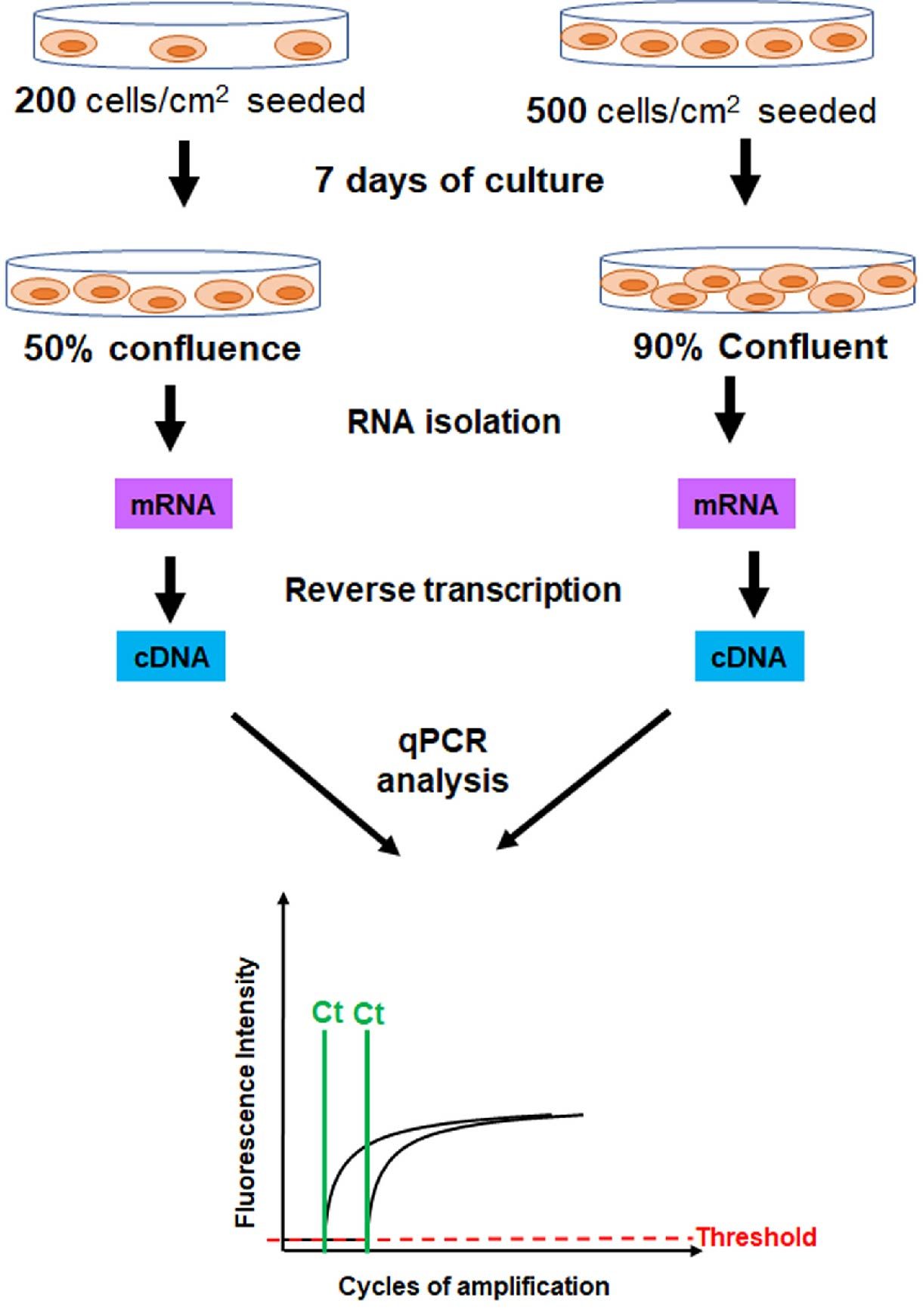
Similar findings were reported in a different study by Kim et al, in which they evaluated the gene expression profiles of mesenchymal stem cells (MSCs) plated at different densities. Adipose tissue-derived MSCs (AT-MSCs) were seeded at a density of 200 or 5,000 cells/cm2 then cultured for 7 days. At the end of 7 days, cells were harvested, total RNA isolated, and the expression level of stemness genes was evaluated by RT-qPCR (Figure 4). The investigators noted that the proliferation rate of AT-MSCs harvested at low density (~50% confluence) was higher than that of AT-MSCs harvested at high density (90% confluence). Importantly, they found that the expression levels of stemness genes, Nanog and c-Myc, were upregulated in AT-MSCs harvested at low density/high proliferation rate compared to AT-MSCs harvested at high density/low proliferation rate (Kim et al., 2017). These results indicated that cell density and proliferation rate at harvesting modulate stemness gene expression.
It is important to note, that in both studies, cells in the experimental and reference groups were treated the same way. They were also maintained in the same culturing conditions (i.e. CO2 levels, humidity, serum levels). The only difference between the two groups was the confluency of the cells at the time of harvest and RNA isolation.
This indicates that if cells are not consistently cultured to reach the same level of confluency at the time of harvest, the gene expression of your reference sample will vary between experiments and as a result be a poor standard for comparison with your experimental sample.
Thus, it is important to control for cell confluency for proper analysis of gene expression data and the generation of gene expression profiles.
-------------
Live-cell imaging is a kinetic method to evaluate cell growth and confluency levels. As such, live-cell imaging can be a powerful tool to normalize measurements of gene expression levels. For this purpose, we recommend the CytoSMART Omni. Whole-well time-lapse imaging and automated image analysis. A fully automated device that works from within cell culture incubators.
--------------
Cell density affects the production of viral vectors
A common application for gene reprogramming is the generation of viral vectors with mammalian cells (Warnock, Daigre and Al-Rubeai, 2011).
Viruses have the capacity to transport their genomes inside the cells they infect. Viral vectors are modified viruses in which the disease-causing genes are removed and replaced with genes that encode a specific desired effect (Chen, Keiser and Davidson, 2018). For example, a lentivirus carrying the gene for the tumor suppressor p53 can be used to overexpress p53 in cancer cells and effectively shut down malignant cell proliferation.
Thus, viral vectors are “carriers” that deliver new genes into target cells, and the reprogrammed target cells utilize this new gene to perform its function.
The production of viral vectors requires two phases (Figure 5). The first phase includes the production of the plasmids that encode helper virus functions (e.g. envelope and gag-polymerase genes) and the gene of interest.
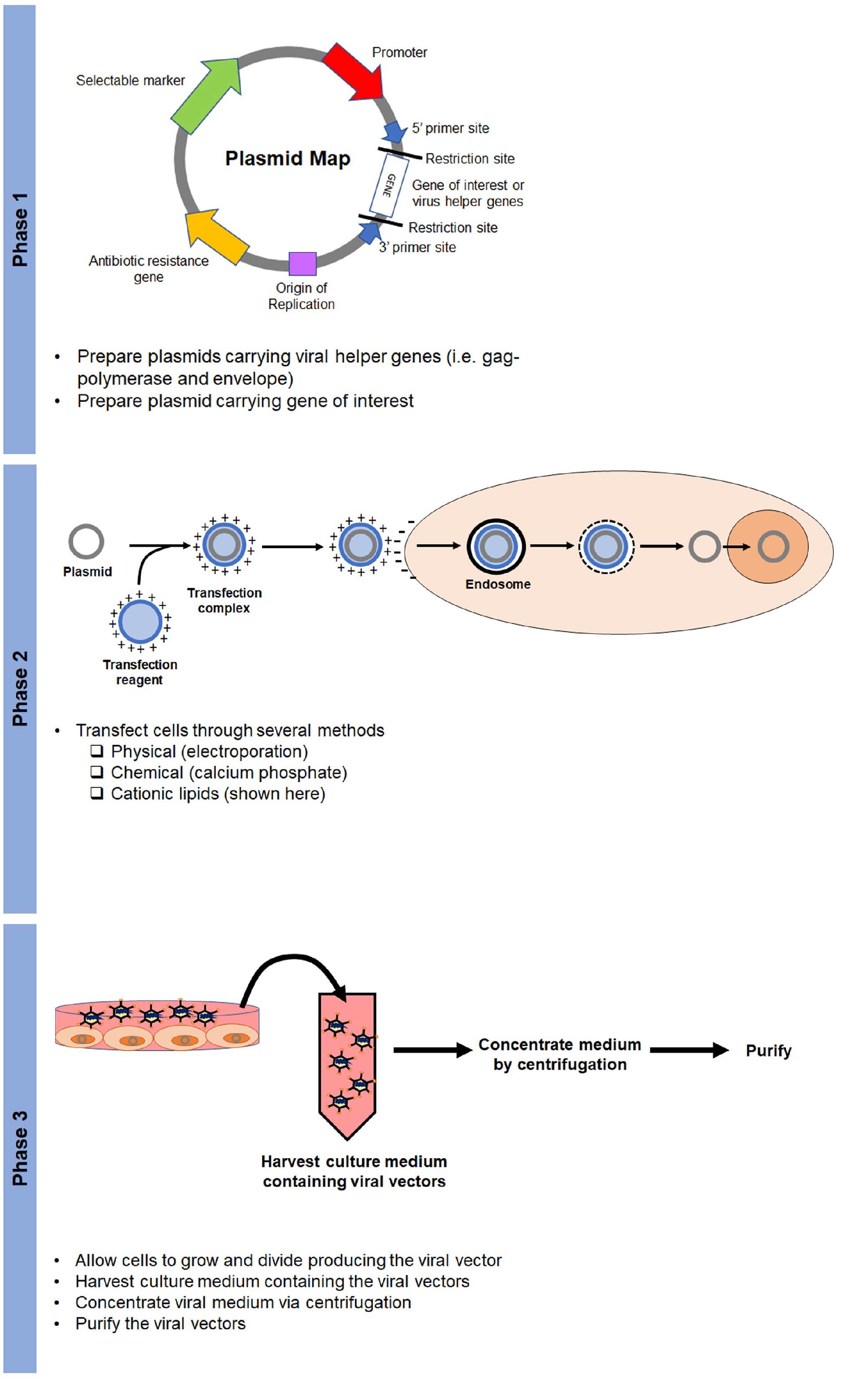
The second phase is cell transfection. Transfection is the process by which foreign DNA is introduced to target cells. This can be done mechanically (electroporation), chemically (calcium phosphate), or with cationic lipids (lipofectamine). Once cells are transfected with plasmids, they will generate the viral vector, which is then harvested, concentrated, and purified (phase 3) (Figure 5).
Transfection is the limiting step. If cells are not properly transfected, the viral yield will be dramatically low.
Not surprisingly, cell confluency greatly influences transfection efficiency. Most transfection protocols recommend transfecting cells at 40-80% confluency. This is because actively dividing cells take up foreign DNA better than quiescent cells, and over-confluent (more than 80%) cells undergo contact inhibition and eventually enter quiescence.
In other words, at optimal confluency, cells will readily take up foreign DNA and undergo genetic reprogramming to consequently yield the desired product. In contrast, at suboptimal confluency, cells will not be easily transfected and not manufacture the desired product.
This phenomenon was demonstrated by the aforementioned study by Han et al (Han et al., 2006). In addition to evaluating the differences in gene expression between sub- and over-confluent cells, the investigators measured the number of adenoviral vectors produced by HEK293T cells cultured at different states of confluence. They found that the yield of adenoviral vectors was significantly lower in over-confluent cells compared to 40 or 90% confluent cells (Figure 6).
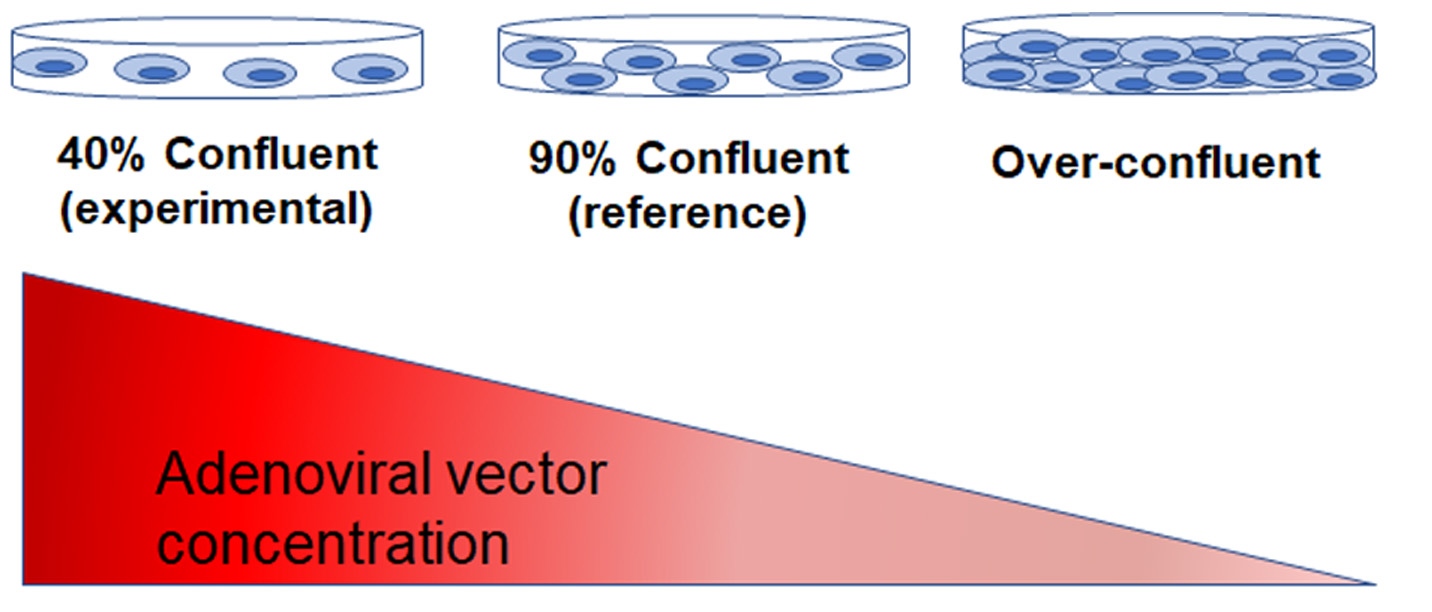
How did this happen?
At 90% confluency, the majority of HEK293T cells are quiescent; thus, are less able to uptake foreign nucleic acids. Since the transfection efficiency is dramatically decreased, the cells are not sufficiently reprogrammed, and the amount of viral yield is reduced. When over-confluent, the number of dividing cells dwindles even more, resulting in subpar transfection efficiency and viral yield.
What does this mean?
Any large-scale manufacture of gene products, like viral vectors, requires that each batch is consistent in quality and quantity. This means that there needs to be very little to no variability in the status of the cells that generate these products.
In sum, controlling for confluency is critical for reliable and robust manufacture of cell products.
----------
For real-time insight into confluency levels of cell cultures without having to enter the lab we recommend the CytoSMART Lux2 Duo Kit. Integrated confluency analysis based on brightfield imaging helps researchers prevent overgrowing cell cultures and helps to determine the ideal moment to perform transfection experiments.
-----------
How can we control for cell confluency in gene expression or gene reprogramming studies?
Now that we know how influential cell confluency is in gene expression profiling and gene reprogramming, how can make sure that we are controlling for it when designing our studies?
The cheapest and fastest method to determine confluency is the examination by eye. Unfortunately, this method is highly susceptible to user bias and introduces variables that cannot be well documented or controlled. For example, what one person views as “over-confluent” may not be considered over-confluent by another.
A better alternative is making assumptions based on seeding density and total RNA yield. However, these methods also have limitations.
First, the doubling time of each cell line is different, and an assumption made for one cell line may not be applicable for another. If one cell line has a doubling time of 12 hours, while another 36 hours, at the end of a 3-day experiment, one cell line would have doubled 6 times while another only twice. Let’s apply this to a gene reprogramming study. Since dividing cells uptake exogenous DNA better than non-dividing cells, the transfection efficiency would be significantly better in one cell line than another, and the viral yield would be different as well. So, without knowing the doubling time of each type of cell, controlling for seeding density alone would be insufficient for consistent transfection.
Second, despite lysing equal numbers of cells, different RNA isolation protocols will yield different amounts of total RNA. So, if you “control” for cell confluency by RNA yield, you must ensure that the protocol utilized to isolate RNA is the same every single time.
Thus, in order to properly control for cell confluency, we need a tool that provides us with quantifiable, real-time information regarding the status of the cell without having to harvest and lyse cells.
Luckily, recent developments in machine learning and live-cell imaging have greatly improved our ability to evaluate cell confluency. Perhaps, soon, all labs will utilize these updated technologies to improve the reliability and reproducibility of gene expression studies.
Related Products
There are currently no products tagged to this resource.